Fibrosi cistica
Revisione paritaria di Dr Hayley Willacy, FRCGP Ultimo aggiornamento di Dr Toni Hazell, MRCGPUltimo aggiornamento 16 maggio 2023
Rispetta le linee guida editoriali
- ScaricaScarica
- Condividi
- Language
- Discussione
- Versione audio
- Aggiungi alle fonti preferite su Google
In questa serie:Test del sudore
La fibrosi cistica è una malattia ereditaria che colpisce principalmente i polmoni e il pancreas, ma può coinvolgere altri organi. I sintomi di solito iniziano in tenera età e includono tosse persistente, sibili, infezioni ricorrenti al torace, difficoltà nella digestione del cibo e malessere generale.
I trattamenti includono antibiotici, fisioterapia, farmaci per fluidificare il muco, sostituti degli enzimi pancreatici e altre terapie.
A colpo d'occhio
Cystic fibrosis is a genetic condition affecting mainly the lungs and pancreas.
It causes cells to make thicker than normal mucus and secretions.
Symptoms often appear in the first year of life, but severity varies.
Common symptoms include persistent cough, wheezing, and recurring chest infections.
Digestive issues can lead to poor growth and large, smelly stools.
There is no cure, but treatments help manage symptoms and improve outlook.
Cos'è la fibrosi cistica?
La fibrosi cistica è una condizione che colpisce principalmente i polmoni e il pancreas, ma può interessare anche altre parti del corpo, tra cui il fegato, il naso, i seni e le ghiandole sudoripare. Normalmente, le cellule di queste parti del corpo producono muco e altri liquidi e secrezioni acquose.
Nei persone con fibrosi cistica, queste cellule non funzionano correttamente e producono muco e secrezioni più dense del normale. Ciò può causare vari sintomi e problemi (che sono descritti di seguito).
Sintomi della fibrosi cistica
I sintomi della fibrosi cistica di solito si manifestano per la prima volta entro il primo anno di vita, ma potrebbero non apparire fino alla tarda infanzia. La gravità dei sintomi può variare.
Sintomi polmonari
Le cellule che rivestono le vie aeree dei polmoni producono muco (catarro) più denso del normale, che non viene facilmente eliminato dai polmoni. Questo può intrappolare i germi (batteri) nelle piccole vie aeree e portare a infezioni e infiammazioni. Quindi, i sintomi che si sviluppano tipicamente includono:
Tosse persistente che di solito produce molta escreato.
Respiro corto e difficoltà respiratorie.
Recurring infezioni toraciche. These can be severe, such as polmonite. Infezioni e infiammazioni ripetute possono danneggiare i polmoni e portare a una scarsa funzione polmonare.
Sintomi intestinali (sistema digestivo)
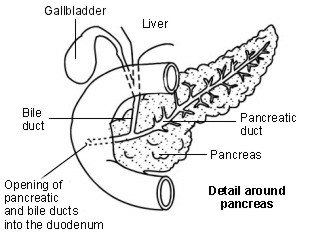
Il pancreas produce normalmente succhi digestivi che contengono sostanze chimiche (enzimi). I succhi digestivi normalmente scorrono dal dotto pancreatico nel duodeno e digeriscono il cibo.
Diagramma che mostra i dettagli intorno al pancreas
Funzione del fegato
Nei persone con fibrosi cistica, le secrezioni ispessite bloccano il normale flusso dei succhi digestivi dal pancreas. Ciò può causare una cattiva digestione o assorbimento del cibo, in particolare dei cibi grassi e delle vitamine liposolubili (vitamine A, D, E e K).
Questo può provocare:
Malnutrizione che porta a una crescita lenta e a un aumento di peso insufficiente (anche se hai un buon appetito e mangi molto, poiché il problema riguarda la digestione e l'assorbimento del cibo).
Le feci grandi, maleodoranti, grasse e oleose si verificano in circa un terzo dei casi.
Addome gonfio.
In circa 3 casi su 10, il pancreas funziona bene e non ci sono, o sono minimi, sintomi intestinali e principalmente sintomi polmonari.
Talvolta i sintomi si manifestano alla nascita
Circa 1 bambino su 10 con fibrosi cistica viene diagnosticato poco dopo la nascita. Ciò è dovuto a una condizione chiamata ileo meconiale, in cui in alcuni casi l'intestino si ostruisce con il meconio. Il meconio è una sostanza densa, scura e appiccicosa prodotta dall'intestino del bambino prima della nascita. Potrebbe essere necessario un intervento chirurgico urgente per alleviare l'ostruzione.
Altri sintomi e complicazioni
Altri organi potrebbero essere colpiti, causando in alcuni casi vari altri problemi. Inoltre, il pancreas e le vie respiratorie potrebbero essere gravemente interessati. Pertanto, altri problemi che possono verificarsi in alcuni casi includono:
Infertilità (especially in males, as the tubes which carry the sperm can become blocked).
Danno al fegato, che può portare a 'cicatrizzazione' del fegato (cirrosi). Il danno al fegato si verifica in circa 1 caso su 12 (se i piccoli dotti nel fegato si ostruiscono o si danneggiano).
Diabete. (Le cellule speciali nel pancreas producono insulina. Se il pancreas si danneggia gravemente nel tempo, i livelli di insulina diminuiscono e può svilupparsi il diabete correlato alla fibrosi cistica.) Questo è raro nei bambini, ma è più comune negli adulti che hanno avuto la fibrosi cistica.
'Assottigliamento' delle ossa (osteoporosi) may develop due to poor absorption of certain foods and, in particular, vitamin D which is needed to maintain healthy bones.
Il sudore ha un sapore molto salato.
Casi lievi
Alcuni casi di fibrosi cistica vengono diagnosticati negli adulti che presentano sintomi relativamente lievi. Ciò potrebbe essere dovuto al fatto che alcuni errori del gene della fibrosi cistica non sono così gravi come altri. La gestione di sodio e cloruro potrebbe essere solo leggermente compromessa in questi casi.
Quali sono le cause della fibrosi cistica?
La fibrosi cistica è un disturbo genetico. Un disturbo genetico è uno che può essere trasmesso dai tuoi genitori attraverso i tuoi geni.
Se hai la fibrosi cistica, uno dei tuoi geni non funziona correttamente. Questo è noto come gene CFTR, che si trova sul cromosoma 7. Possono verificarsi diversi errori in questo gene e ciò significa che ci sono diverse gravità della fibrosi cistica.
Il gene CFTR aiuta a controllare il modo in cui le cellule gestiscono il sale (ioni di sodio e cloruro). L'errore nel gene provoca l'incapacità delle cellule di gestire correttamente sodio e cloruro.
Di conseguenza, le cellule negli organi interessati presentano un difetto nel modo in cui sodio e cloruro si spostano dentro e fuori dalle cellule. Fondamentalmente, troppo sodio entra nelle cellule. L'acqua segue il sodio, lasciando troppo poca acqua all'esterno delle cellule. Questo provoca un muco o secrezioni acquose all'esterno delle cellule troppo dense (ad esempio, nelle vie respiratorie dei polmoni).
Come si eredita la fibrosi cistica?
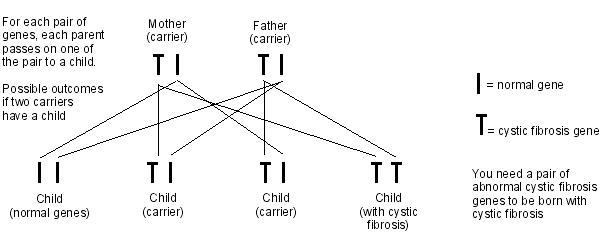
La fibrosi cistica è una malattia autosomica recessiva. Ciò significa che per sviluppare la fibrosi cistica è necessario ereditare due geni della fibrosi cistica, uno dalla madre e uno dal padre. Se erediti solo un gene della fibrosi cistica, sei chiamato portatore.
Circa 1 persona su 25 nel Regno Unito di discendenza europea bianca è portatrice del gene della fibrosi cistica. È molto meno comune nelle persone afro-caribiche e asiatiche. I portatori non hanno la malattia, poiché possiedono un gene normale che può controllare il trasporto del sale nelle loro cellule. Ma i portatori possono trasmettere il gene della fibrosi cistica ai loro figli.
Quando due persone portano il gene della fibrosi cistica e hanno un figlio, c'è un:
1 su 4 possibilità che il bambino abbia la fibrosi cistica (ereditando il gene della fibrosi cistica da entrambi i genitori).
2 su 4 probabilità che il bambino non abbia la fibrosi cistica ma sia portatore (ereditando un gene per la fibrosi cistica da un genitore e il gene normale dall'altro genitore).
1 su 4 probabilità che il bambino non abbia la fibrosi cistica e non sia portatore (ereditando il gene normale da entrambi i genitori).
Diagramma per mostrare l'eredità della fibrosi cistica
Quanto è comune la fibrosi cistica?
Circa 1 bambino su 2.500 nel Regno Unito nasce con fibrosi cistica. Attualmente, oltre 9.000 persone nel Regno Unito sono affette da fibrosi cistica.
Cystic fibrosis diagnosis
Test del sudore
Un medico può prescrivere un test del sudore se sospetta la fibrosi cistica dai sintomi. Questo test misura la quantità di sale (sodio e cloruro) nel sudore della pelle. Le persone con fibrosi cistica hanno un livello di sale insolitamente alto nel sudore.
Test genetico
Un test genetico può confermare la diagnosi. Alcune cellule vengono raschiate dall'interno della guancia o prelevate da un test del sangue. Questi possono essere analizzati per rilevare il gene della fibrosi cistica.
Test di screening
Tutti i neonati nel Regno Unito vengono ora sottoposti a screening per la fibrosi cistica .Viene eseguito un piccolo prelievo di sangue dal tallone circa il sesto giorno dopo la nascita. Questo può rilevare una sostanza chimica chiamata tripsinogeno immunoreattivo, che è elevata nei neonati con fibrosi cistica. Se il livello è alto, si può effettuare un test del sudore e un test genetico per confermare la diagnosi. Lo screening è considerato importante perché prima viene fatta la diagnosi, prima può iniziare il trattamento, migliorando così la prognosi.
Altri test
Other tests may be performed to check for the development of complications. These may include radiografie, ecografie e rimozione di una piccola quantità di tessuto corporeo (una biopsia) to examine under a microscope.
Trattamento della fibrosi cistica
Ci sono molti aspetti nel trattamento delle persone con fibrosi cistica. Il trattamento coinvolge il contributo, i consigli e l'expertise di vari professionisti. Questi includono medici della salute infantile, infermieri specialistici, fisioterapisti, dietisti, consulenti e psicologi, oltre al tuo team di assistenza primaria.
È consuetudine effettuare controlli e test regolari per monitorare la condizione e tenere sotto controllo la crescita, lo sviluppo e il benessere dei bambini.
Di seguito è riportata una panoramica dei trattamenti più comunemente usati. Tuttavia, non si tratta di un elenco completo o esaustivo di tutti i trattamenti impiegati. È necessario un piano di trattamento individuale per ogni caso, tenendo conto delle circostanze specifiche.
TRATTAMENTI PER I PROBLEMI POLMONARI
Physiotherapy and exercise
La fisioterapia toracica regolare è molto importante. Aiuta a liberare le vie respiratorie dal muco spesso e appiccicoso. Di solito, un fisioterapista mostra ai genitori come farlo per i loro bambini. Consiste in un modo speciale di battere fermamente sul petto mentre il bambino è sdraiato con la testa rivolta verso il basso per favorire l'espulsione del catarro. La fisioterapia toracica due volte al giorno è una pratica comune. Potrebbe essere necessario aumentare la frequenza durante i periodi di infezioni respiratorie.
Esiste una vasta gamma di tecniche di clearance delle vie aeree disponibili e il tuo fisioterapista sarà in grado di consigliarti quella o quelle più adatte a te. Tuttavia, queste potrebbero cambiare man mano che invecchi o che la tua condizione si modifica nel tempo.
È anche importante incoraggiare i bambini a fare esercizio e a essere il più attivi e in forma possibile. Pertanto, si promuovono sport e giochi come corsa, nuoto, calcio e tennis.
Antibiotici e antifungini
Corsi di antibiotici are a mainstay of treatment. Many children with cystic fibrosis take regular long-term antibiotics. The dose is increased and/or other types of antibiotics are given when a chest infection develops. Various germs (bacteria) can cause infections and the antibiotics chosen depend on which bacteria are found in samples of sputum.
Gli antibiotici somministrati per via endovenosa (intravenosa) sono spesso necessari per infezioni gravi che non rispondono alle compresse di antibiotici. Possono anche essere somministrati tramite un nebulizzatore. Un nebulizzatore permette di inalare un farmaco come una nebbia fine, attraverso una maschera.
A bacterium called Pseudomonas aeruginosa commonly persists in the thick mucus in the airways. To keep this from flaring up into repeated infections, an antibiotic given by nebuliser or inhaler is a common treatment.
Qualche volta i polmoni si infettano con un fungo e è necessario un farmaco antifungino.
Inalatori
Inhalers to open up the airways as much as possible may be used - for example, salbutamolo. Questo è simile al trattamento usato per l'asma.
Dornase alfa
Questo è un medicinale somministrato tramite nebulizzatore in alcuni casi. Aiuta a rompere e a fluidificare il muco denso, rendendo più facile tossire e liberare le vie respiratorie dal muco. Potrebbe ridurre il numero di infezioni polmonari e contribuire a migliorare la funzione polmonare.
Ossigeno
Le persone con malattie polmonari avanzate potrebbero beneficiare dell'ossigeno, soprattutto durante la notte.
Other medication to improve lung function - for example, ibuprofene e azitromicina - may also be recommended in some cases.
TRATTAMENTI PER I PROBLEMI PANCREATICI
Nutrizione
Le sostanze chimiche (enzimi) necessarie per digerire il cibo sono notevolmente ridotte nella maggior parte delle persone con fibrosi cistica. Pertanto, i bambini con fibrosi cistica devono seguire una dieta ricca di grassi e carboidrati. Di solito, un dietista fornirà consigli dettagliati. Potrebbero essere necessari anche integratori di bevande ad alto contenuto energetico. Inoltre, sono necessari integratori vitaminici, poiché molte vitamine presenti negli alimenti non vengono assorbite molto bene. Essere ben nutriti ti aiuterà anche a combattere eventuali infezioni polmonari.
Integratori di enzimi
Nella maggior parte dei casi, sono necessari integratori di enzimi per aiutare la digestione del cibo. (Questi sostituiscono gli enzimi che normalmente provengono dal pancreas.) È necessario assumere questi integratori ogni volta che si mangia. Ciò può significare prendere molte dosi al giorno.
ALTRI TRATTAMENTI
In alcuni casi, possono svilupparsi altri problemi correlati alla fibrosi cistica che richiedono trattamento. Ad esempio:
La carenza di sale può verificarsi in condizioni di caldo e potrebbe richiedere integratori di sale.
In alcuni casi si sviluppano problemi al fegato e potrebbe essere necessario un trattamento specialistico per il fegato.
Se si sviluppa il diabete, di solito è necessario un trattamento con insulina o compresse.
Talvolta si sviluppano escrescenze nasali (polipi) and can be treated with steroid nasal drops and sprays.
Reflusso acido dallo stomaco all'esofago is common and can be treated with medicines which reduce the acid content of the stomach juices.
La stitichezza è abbastanza comune e può richiedere l'uso regolare di lassativi.
All people with cystic fibrosis should be up to date with vaccinazioni di routine and have an vaccino antinfluenzale annuale per prevenire l'influenza. It is also important to be immunised with vaccino pneumococcico to help prevent pneumonia caused by this bacterium and people with cystic fibrosis were eligible for the COVID-19 vaccination, but are not included in the 2023 Spring booster campaign. It remains to be seen if ongoing COVID-19 vaccination will be offered to this group.
Alcuni bambini vengono sottoposti a test per verificare la loro immunità contro il varicella (varicella). Se non hai immunità, potresti ricevere la vaccinazione contro varicella.
In alcuni casi, potrebbe essere offerto un trapianto di polmone o di cuore/polmone man mano che la malattia polmonare peggiora.
Stanno venendo studiati e sviluppati trattamenti più recenti e, se si dimostrano efficaci, potrebbero diventare più diffusi in futuro. Per esempio:
Terapia genica. Questo comporta l'uso di uno spray inalato per consegnare copie normali del gene della fibrosi cistica ai polmoni.
Si stanno testando farmaci che potrebbero correggere il regolamento anomalo di sale e acqua delle cellule, che porta alla produzione di muco denso e secrezioni nei polmoni e in altri organi.
Nuovi metodi per migliorare l'efficacia dei trattamenti attuali sono in fase di sviluppo.
Qual è la prospettiva (prognosi)?
I trattamenti per la fibrosi cistica sono migliorati notevolmente negli ultimi decenni. Tuttavia, si tratta di una condizione che dura tutta la vita e attualmente non esiste una cura per la fibrosi cistica.
Ci saranno momenti in cui i sintomi saranno più gravi - principalmente quando si sviluppa un'infezione polmonare. Anche con il trattamento, i principali rischi sono le infezioni ricorrenti ai polmoni e la polmonite. Questo può avere un effetto dannoso ripetuto sulla funzione polmonare, che può peggiorare nel tempo. La maggior parte delle persone con fibrosi cistica muore a causa di complicanze polmonari, principalmente insufficienza respiratoria e cardiaca.
Speranza di vita nella fibrosi cistica
Con un trattamento migliorato, si è registrato un aumento drastico della sopravvivenza delle persone con fibrosi cistica nel corso di circa 20 anni.
Negli anni '60 e prima, la maggior parte dei bambini nati con fibrosi cistica sopravviveva solo pochi mesi o anni. Oggi, molte persone con fibrosi cistica vivono fino alla fine dei loro 30 anni e oltre. Con cure e trattamenti ottimali, si stima che oltre la metà dei bambini con fibrosi cistica di oggi dovrebbe vivere fino ai loro 40 o 50 anni. Con il trattamento, la maggior parte delle persone con fibrosi cistica può vivere una vita ragionevolmente normale e produttiva.
Tuttavia, la morte in età infantile o in età adulta precoce non è ancora rara.
Consulenza genetica per la fibrosi cistica
People with a family history of cystic fibrosis may wish to have Consulenza genetica e test to find out their risk of passing the condition on to their children. A simple test can be done to look at the genes from cells from the inside of the cheek or from blood. The test can detect the cystic fibrosis gene which can show if you are a carrier of the abnormal gene.
Scelte del paziente per Condizioni genetiche

Salute dei bambini
Sindrome di Turner
La sindrome di Turner è una condizione genetica che colpisce solo le ragazze. Le caratteristiche più distintive della sindrome sono essere basse di statura, avere alcune caratteristiche fisiche (dettagliate di seguito) e ovaie che non funzionano correttamente. Sebbene non esista una cura, ci sono trattamenti che possono aiutare la maggior parte delle ragazze con la sindrome di Turner a condurre una vita relativamente normale.
di Dr Colin Tidy, MRCGP

Salute dei bambini
Cardiopatia congenita
Le malattie cardiache congenite sono condizioni in cui si sviluppa un'anomalia (difetto) nel cuore prima della nascita. Esistono diversi tipi di difetti cardiaci congeniti. Alcuni sono lievi e causano pochi problemi; altri sono potenzialmente letali per il bambino.
di Dr Mary Harding, MRCGP
Domande frequenti
What happens if the pancreas or airways become severely affected by cystic fibrosis?
If the pancreas and airways become severely affected by cystic fibrosis, other problems can develop. These may include repeated sinus infections, growths (polyps) in the nose, infertility (especially in males due to blocked tubes carrying sperm), liver damage leading to scarring (cirrhosis), diabetes, inflammation of the pancreas (pancreatitis), and rectal prolapse. Bone thinning (osteoporosis) can also occur due to poor absorption of vitamin D.
Can cystic fibrosis be diagnosed in adults even if symptoms are mild?
Yes, some cases of cystic fibrosis are diagnosed in adults who have relatively mild symptoms. This can happen if the errors in the cystic fibrosis gene are not as severe as others, leading to only a mild effect on how the body handles sodium and chloride.
How often do individuals with cystic fibrosis need to take enzyme supplements?
Most people with cystic fibrosis need to take enzyme supplements every time they eat food. This can mean taking many doses throughout the day to help digest food, as their pancreas may not produce enough of the necessary digestive chemicals.
Are there any specific vaccines recommended for people with cystic fibrosis?
Yes, it is important for people with cystic fibrosis to be up-to-date with routine immunisations. They should also have an annual flu jab and be immunised with the pneumococcal vaccine to help prevent pneumonia. Additionally, some children are tested for chickenpox immunity, and if they lack it, they may be offered the chickenpox vaccine.
What kind of specialists are involved in treating cystic fibrosis?
The treatment for cystic fibrosis involves a team of various professionals. These include child health doctors, specialist nurses, physiotherapists, dieticians, counsellors, psychologists, and the primary healthcare team. An individual treatment plan is created for each person to address their specific needs.
What new treatments are being explored for cystic fibrosis?
New treatments for cystic fibrosis are being researched and developed. These include gene therapy, which uses an inhaled spray to deliver normal copies of the cystic fibrosis gene to the lungs. Also, medicines are being tested that aim to correct the abnormal salt and water regulation in cells, which causes thick mucus. Additionally, new methods to enhance the effectiveness of existing treatments are being developed.
Ulteriori letture e riferimenti
- Standard di assistenza; Fondazione Fibrosi Cistica
- Sodio colistimetato e tobramicina in polvere secca per inalazione per il trattamento dell'infezione polmonare da Pseudomonas nella fibrosi cistica; Linee guida di valutazione tecnologica NICE, marzo 2013
- Conway S, Balfour-Lynn IM, De Rijcke K, et al; Standard della Società Europea di Fibrosi Cistica: Quadro di riferimento per il Centro di Fibrosi Cistica. J Cyst Fibros. Maggio 2014;13 Suppl 1:S3-22. doi: 10.1016/j.jcf.2014.03.009.
- Fibrosi cistica: diagnosi e trattamento; Linee guida NICE (ott 2017)
- Langton Hewer SC, Smyth AR; Strategie antibiotiche per l'eradicazione di Pseudomonas aeruginosa nelle persone con fibrosi cistica. Cochrane Database Syst Rev. 2017 Apr 25;4:CD004197. doi: 10.1002/14651858.CD004197.pub5.
- Lynch JP 3°, Sayah DM, Belperio JA, et al; Trapianto di polmone per fibrosi cistica: risultati, indicazioni, complicanze e controversie. Semin Respir Crit Care Med. Apr 2015;36(2):299-320. doi: 10.1055/s-0035-1547347. Epub 2015 Mar 31.
Informazioni sull'autoreVisualizza il profilo completo

Dr Toni Hazell, MRCGP
MBBS, BSc, MRCGP, DFSRH, Dip GU med, DRCOG, DCH (London, UK, 2000)
La Dott.ssa Toni Hazell si è laureata presso la St. Mary’s Hospital Medical School e ha completato il suo VTS al Northwick Park Hospital.
Informazioni sul recensoreVisualizza il profilo completo

Dr Hayley Willacy, FRCGP
Medico di base, Autore medico
MBChB (1992), DRCOG, DFFP, MRCOG (Part 1) MRCGP (2007), DFSRH (2013), MSc - medical education (2020)
La Dott.ssa Hayley Willacy era un medico di base del NHS che lavorava nel nord-ovest dell'Inghilterra, e si è ritirata dalla pratica clinica nel 2022 dopo 30 anni.
Storia dell'articolo
Le informazioni su questa pagina sono scritte e revisionate da clinici qualificati.
Articolo disponibile anche in Inglese, Tedesco, Spagnolo, Francese, Italiano, Portoghese, Hindi, Ebraico, Arabo, and Svedese.
Prossima revisione prevista: 12 maggio 2028
16 maggio 2023 | Ultima versione

Chiedi, condividi, connettiti.
Esplora le discussioni, fai domande e condividi esperienze su centinaia di argomenti di salute.

Non ti senti bene?
Valuta i tuoi sintomi online gratuitamente
Iscriviti alla newsletter di Patient
La tua dose settimanale di consigli sulla salute chiari e affidabili - scritti per aiutarti a sentirti informato, sicuro e in controllo.
Abbonandoti accetti i nostri Informativa sulla Privacy. Puoi annullare l'iscrizione in qualsiasi momento. Non vendiamo mai i tuoi dati.
Di più sulla salute dei bambini
- Enuresi notturna
- Medicina per l'enuresi notturna - desmopressina
- Sistemi di ricompensa per l'enuresi notturna
- Tumori infantili
- Piede torto
- Tosse e raffreddori nei bambini
- Sindrome di Down
- Intossicazione alimentare nei bambini
- Gastroenterite nei bambini
- Malattia di Hirschsprung
- Intussuscezione e volvolo nei bambini
- Dermatite da pannolino
- Polio e vaccino antipolio
- Rosolia infantile
- Rotavirus
- Strabismo nei bambini
- Tetano e il vaccino antitetanico
- Diarrea del bambino
- Infezione urinaria nei bambini
- Tumore di Wilms