Huntington's disease
Peer reviewed by Dr Colin Tidy, MRCGPLast updated by Dr Toni Hazell, MRCGPLast updated 21 Dec 2023
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Huntington's disease is a genetically inherited condition that causes parts of the brain and nervous system to stop working properly over time. It is a slowly progressive condition that interferes with the movements of your body, thinking and judgement, and can lead to a change in your behaviour.
The symptoms occur because of damage and death of some of the brain cells (neurons) in particular parts of your brain. Genetic testing helps to diagnose Huntington's disease.
At present there is no cure for Huntington's disease. Treatment is aimed at trying to control symptoms as much as possible when they develop.
At a glance
Huntington's disease is a genetic condition that damages brain function and worsens over time.
It can cause problems with movement, thinking, judgement, and behaviour.
Symptoms often appear between the ages of 30 and 50 but can occur earlier.
There is no cure, but treatments help manage the symptoms.
If you have neurological symptoms, especially with a family history of Huntington's disease, see a doctor.
What is Huntington's disease?
Huntington's disease is a genetically inherited condition that gradually damages brain function. It progresses over time, often proving fatal by the age of 40 - 50.
It can lead to a change in your behaviour and interfere with:
Movements of your body.
Reasoning.
Awareness.
Thinking.
Judgement (cognition).
The disease was named after George Huntington who first described it in 1872.
How common is Huntington's disease?
Huntington's disease affects between 11 - 14 people per 100,000 in the UK, the rest of Europe and North America. Worldwide, it seems to be more common amongst white populations than amongst Asian or African people. Huntington's disease affects both men and women equally.
How common is juvenile Huntington's disease?
Between 5-10 in 100 people with Huntington's disease develop symptoms before they are 20. This is known as juvenile Huntington's disease.
It can cause more severe symptoms. Most people who have this type of Huntington's disease inherit the condition from their father.
What are the symptoms of Huntington's disease?
The symptoms of Huntington's disease can vary from person to person, and gradually worsen over time. Symptoms can develop at any time, but they tend to emerge between the age of 30 and 50.
Symptoms are grouped into three areas:
Movement.
Cognitive.
Mood and behavioural problems.
Movement problems
Huntington's disease can cause both involuntary and impaired movement problems, which include:
Jerky, involuntary movements in the head, face, arms or legs (chorea).
Muscle spasms mostly in the shoulders, neck, arms and legs (dystonia).
Stiff or rigid limbs and slower movements (bradykinesia).
Difficulty swallowing.
Slurred speech.
Slow eye movements.
In severe chorea, you can develop uncontrollable movements of your arms or legs (called ballism). This can interfere with your ability to move around. You may be more likely to fall, have difficulty holding things, and feeding or dressing yourself.
As Huntington's disease progresses, chorea symptoms tend to gradually be replaced by dystonia. Dystonia can lead to twisting movements, repetitive movements, or abnormal postures.
Because the disease can also affect the muscles that control your swallowing, choking can be a problem as well as weight loss due to difficulty eating.
The slow and unusual eye movements can make it difficult looking from side to side or up and down.
Cognitive problems
Because Huntington's disease affects your brain as well as your nervous system, cognitive problems can develop, these include:
Short-term memory loss and memory lapses.
Lack of concentration; not being able to focus, organise, or prioritise tasks.
Trouble processing thoughts.
Orientation problems.
Difficulty learning new skills or information.
Impaired judgment.
Lack of impulse control.
The slow decline in cognition can be similar to a dementia-type problem.
Mood and behavioural problems
Changes in your behaviour can be one of the first signs of Huntington's disease and they may come on before your movement is affected. These include:
Irritability, apathy or sadness.
Low interest in self-care; for example, washing less frequently, not taking care of your appearance, and being untidy.
Mental health problems including depression or suicidal thoughts.
Depression is the common psychological symptom in people with Huntington's disease and there is an increased risk of suicide.
Other mental health problems include:
The personality changes related to Huntington's disease can be very difficult for you or your loved ones to deal with, and can put a strain on your relationships.
You may find it difficult to accept that your behaviour may be a problem. It is all part of the way that Huntington's disease affects your brain
Symptoms of juvenile Huntington's disease
Juvenile Huntington's disease is less common than the form which presents later. Symptoms begin in childhood or adolescence (before the age of 20). Around 5 - 10% of all cases of Huntington's disease present at this age. Symptoms are similar to Huntington's disease which presents as an adult, they just occur earlier.
When to see a doctor
If you are experiencing any sort of neurological symptoms then it is important to see a doctor - there are a wide variety of possible causes, including Huntington's disease and other causes. If you know that you have a family history of Huntington's disease, make sure that you mention that at the appointment.
What causes Huntington's disease?
Huntington's disease is caused by a defective gene that you inherit from your parents.
Huntington's disease is an autosomal dominant condition, meaning you can inherit the disease from just one of your parents.
If one of your parents has a faulty copy of the gene, there is a 50:50 chance that each child they have will inherit the faulty gene.
If a member of your family who is carrying the faulty gene dies before their symptoms develop (and therefore before Huntington's disease is diagnosed), relatives will not be aware of the family history.
How Huntington's disease affects the brain
The genetic defect in Huntington's disease means that certain proteins needed to make brain chemicals cannot be made in your brain as normal. It is thought that this leads to damage and death of some of the brain cells in parts of your brain called the basal ganglia and the cortex.
It is this damage that leads to the symptoms of Huntington's disease. There is also a build-up of a chemical called dopamine in the brain which contributes to the problems with moving.
New mutations
Occasionally, someone with Huntington's disease may not have a history of the disease in their family. This may be because of what is called a 'new mutation'.
A new mutation is a fault in a gene that is present for the first time in one family member. It can happen because of:
A fault in the genetic material in either the egg or sperm of one of the affected person's parents.
A fault in the genetic material of the embryo.
It is not clear what causes this mutation.
How is Huntington's disease diagnosed?
Genetic testing is used to confirm whether you carry the faulty gene that causes Huntington's disease.
It is recommended that you undergo counselling if you are considering genetic testing. Your GP will be able to refer you to a specialist who is a genetic counsellor.
Other tests
CT scan or MRI scan of your brain may show some typical signs of Huntington's disease. However, these scans are not usually helpful in the early stages as changes may not be present. Scanning is mostly used in the later stages of the disease.
What is the treatment for Huntington's disease?
At present there is no cure for Huntington's disease. Also, no treatment has been found to delay the onset or progression of symptoms. So, treatment is aimed at trying to control symptoms as much as possible when they do develop. These include:
Medications for physical problems.
Medications for psychological problems.
Physiotherapy.
Occupational therapy.
Speech therapy.
Diet advice.
Counselling, advice and support
Medications for physical problems
There are a number of different medicines that can be used to help treat physical problems. These include:
Benzodiazepines, for example, clonazepam and diazepam to manage chorea.
Tetrabenazine may also be used.
Dopamine agonists and levodopa to manage bradykinesia.
Whether and when to start treatment will depend on striking a balance between the benefits and side-effects.
Medications for psychological problems
If you develop depression, antidepressant medicines can be helpful. Medicines are also available to treat some other mental health problems that may be associated with Huntington's disease.
Physiotherapy
You may be referred to a physiotherapist for help with exercises for your balance and exercises to help you move around more easily.
Occupational therapy
An occupational therapist may be able to help you with any adaptations that you need to make your day-to-day life easier. For example, they can help with adaptations to your home such as wheelchair access, rails and changes to your bedroom and bathroom.
Speech therapy
A speech and language therapist may be able to help with speech and/or swallowing difficulties. They may be able to teach you different ways of communicating.
Dietary advice
You may be referred to a dietician if you lose a lot of weight (due to swallowing difficulties and movement problems). They can advise about foods that may be easier for you to eat because they involve less chewing.
Sometimes swallowing problems mean that a nasogastric tube may be necessary. This is a tube that passes through your nose to your stomach so that food can be delivered to your stomach without you having to swallow.
Other treatments
Most people with Huntington's disease have a team of healthcare specialists who work with them. Your GP often co-ordinates your care. Other team members may include a specialist in brain, nerve and muscle problems (a neurologist), a psychiatrist and a genetic counsellor.
Possible future treatments
Various new treatments for Huntington's disease are being studied. They include gene therapy treatments and various medicines. For example, trials looking at the effect of a new medicine called pridopidine are underway.
Clinical trials are also looking at medicines to prevent people who have the faulty Huntington's disease gene from developing the disease. However, these treatments are still very much at an experimental level.
More research is needed before we will know whether such treatments are helpful for Huntington's disease.
What is the outlook for Huntington's disease?
Huntington's disease slowly progresses and symptoms increase and worsen over time. In the later stages of Huntington's disease, you will become totally dependent on other people and require full-time care. Huntington's disease leads to considerable disability and, at present, will eventually lead to death.
Currently, most people live from between 10 to 25 years after they first develop the symptoms of Huntington's disease. Someone with Huntington's disease usually dies from an infection such as pneumonia but suicide is very common.
Remember that new treatments are under investigation. Your specialist and the Huntington's Disease Association will be able to discuss any treatment developments or trial treatments with you.
Can I pass Huntington's disease on to my children?
If you carry the defective Huntington's disease gene, for each child that you have, there is a 50:50 chance that they will also have Huntington's disease.
If you or your partner have Huntington's disease, prenatal testing is available. This can show whether your baby has the defective gene and therefore whether they will develop the disease. However, testing is not always 100% accurate.
Autosomal dominant inheritance
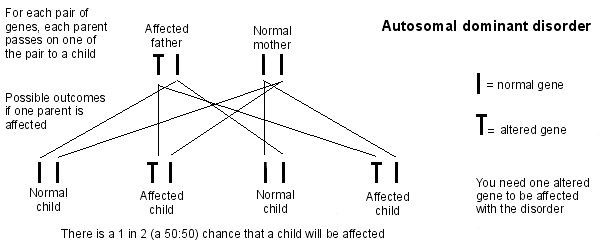
Preimplantation genetic diagnosis (PGD) is also available if one parent carries the defective gene. This basically involves the couple undergoing IVF-type treatment so that embryos can be tested for Huntington's disease before they are implanted in the woman's womb (uterus). Only embryos without the defective Huntington's disease gene are implanted.
Genetic counselling and specialist advice are recommended if you or your partner have Huntington's disease, or if there is Huntington's disease in either of your families, and you are considering pregnancy.
Patient picks for Movement disorders

Brain and nerves
Tourette syndrome
Tourette syndrome is a condition that causes you to make involuntary movements or noises called tics. It tends to be associated with various other problems such as behavioural problems and attention deficit hyperactivity disorder (ADHD). It can often be well managed with psychological treatments and sometimes with medication. Simple motor tics are brief and sudden repetitive movements that involve just a few muscle groups. Simple vocal tics involve saying a word or phrase, or making a different noise. Complex tics involve several different muscle groups in areas throughout your body. Complex motor tics include jumping, bending, twisting or other complex movements. Complex vocal tics include repeating other people’s words or phrases (echolalia), or using obscene, vulgar or swear words (coprolalia). The most common of all childhood tics is Tourette syndrome.
by Dr Colin Tidy, MRCGP

Brain and nerves
Essential tremor
Essential tremor is the term for having uncontrolled shaking movements in parts of your body - most commonly your arms and hands.
by Dr Hayley Willacy, FRCGP
Frequently asked questions
What is the timeline of Huntington's disease from symptom onset to end of life?
Huntington's disease is a progressive condition where symptoms gradually increase and worsen over time. Most people live for 10 to 25 years after they first develop symptoms. In the later stages, individuals become fully dependent on others and require full-time care, eventually leading to death.
What is the average age when symptoms of Huntington's disease typically appear?
Symptoms of Huntington's disease can develop at any time, but they most commonly start between the ages of 30 and 50. In some cases, juvenile Huntington's disease can manifest before the age of 20.
When should I see a doctor if I suspect I might have Huntington's disease?
If you are experiencing any neurological symptoms, it's important to consult a doctor. There are many possible causes for such symptoms. If you have a family history of Huntington's disease, you should definitely mention this to your doctor during the appointment.
How does Huntington's disease progress from chorea to dystonia?
In the early stages of Huntington's disease, an individual might experience chorea, which involves jerky, involuntary movements. As the disease progresses, these chorea symptoms tend to be gradually replaced by dystonia, which can lead to twisting movements, repetitive movements, or abnormal postures.
Are new treatments for Huntington's disease being developed?
Yes, various new treatments for Huntington's disease are currently being studied. These include gene therapy treatments and different medicines. Clinical trials are also investigating medicines aimed at preventing people with the faulty gene from developing the disease. However, these treatments are still experimental, and more research is needed to determine their effectiveness.
What mental health conditions are associated with Huntington's disease?
People with Huntington's disease can experience a range of mental health problems. Depression is a very common psychological symptom, and there is an increased risk of suicide. Other associated conditions include obsessive-compulsive disorder, bipolar disorder, and schizophrenia.
If I have Huntington's disease, how can I ensure my future children won't inherit it?
If you or your partner carry the defective gene, prenatal testing is available to determine if your baby has the gene. Another option is Preimplantation Genetic Diagnosis (PGD), which involves using IVF-like treatment to test embryos for the defective gene before implantation. Only embryos without the faulty gene are implanted. Genetic counselling is recommended to discuss these options and provide specialist advice.
Further reading and references
- Roos RA; Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010 Dec 20;5(1):40. doi: 10.1186/1750-1172-5-40.
- Zielonka D, Mielcarek M, Landwehrmeyer GB; Update on Huntington's disease: advances in care and emerging therapeutic options. Parkinsonism Relat Disord. 2015 Mar;21(3):169-78. doi: 10.1016/j.parkreldis.2014.12.013. Epub 2014 Dec 19.
- European Huntington's Disease Network; Information about the EHDN project and participating studies
- Pandey M, Rajamma U; Huntington's disease: the coming of age. J Genet. 2018 Jul;97(3):649-664.
- Rawlins MD, Wexler NS, Wexler AR, et al; The Prevalence of Huntington's Disease. Neuroepidemiology. 2016;46(2):144-53. doi: 10.1159/000443738. Epub 2016 Jan 30.
- Caron NS, Wright GEB, Hayden MR; Huntington Disease GeneReviews® 1998 Oct 23 [updated 2020 Jun 11].
- Marcus R; What Is Huntington Disease? JAMA. 2023 Sep 12;330(10):1014. doi: 10.1001/jama.2023.13024.
About the authorView full bio

Dr Toni Hazell, MRCGP
MBBS, BSc, MRCGP, DFSRH, Dip GU med, DRCOG, DCH (London, UK, 2000)
Dr. Toni Hazell qualified from St. Mary’s Hospital Medical School and did her VTS at Northwick Park Hospital.
About the reviewerView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 19 Dec 2028
21 Dec 2023 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in brain and nerves
- Carpal tunnel syndrome
- Cauda equina syndrome
- Cluster headaches
- Epilepsy
- Epilepsy medication and side-effects
- Foot drop
- Functional neurological disorder
- Guillain-Barré syndrome
- Medication-overuse headache
- Migraine and combined hormonal contraception
- Migraine treatment
- Postherpetic neuralgia
- Selective mutism
- Shingles
- Stroke
- Sudden unexpected death in epilepsy
- Tension headache
- Tonic-clonic seizures
- Transient ischaemic attack
- Trigeminal neuralgia